Temperature-triggered inflatable hydrogel muscles with snap-through instability for untethered robots – ryan
Since the untethered PAMs require hydrogel actuators possessing tunable elastic modulus as bladders, we designed thermally responsive hydrogels with switchable crosslinking. First, organogels were synthesized by polymerization of octyl acrylate (OA) and acrylamide (AAm) in the presence of β-cyclodextrin (β-CD), with N,N’-methylenebisacrylamide (MBA) and Irgacure 2959 as the crosslinker and initiator, respectively (Fig. 2a). After water exchanging, hybrid cross-linked β-CD/OA hydrogels were obtained, where the hydrogel network included chemical crosslinks, hydrophobic associations of alkyl chains (physical crosslinks), and randomly distributed β-CD. Because the formation of hydrophobic associations in hydrogel networks depended on the content of OA moiety, we investigated the influence of OA molar fraction on the assembly behavior and host-guest interaction between β-CD and P(AAm-co-OA) in the solvents (Fig. S1). When the OA molar fraction of β-CD/P(AAm-co-OA) was 6.7%, a transparent solution formed in water, due to the host-guest interaction between β-CD and OA. In the 1H NMR spectra of β-CD and P(AAm-co-OA), the characteristic peaks of H3 and H5 for β-CD shifted upfield, whereas the characteristic peaks of OA side chains (methyl and methylene groups) on P(AAm-co-OA) shifted downfield, indicating the formation of host-guest interactions between alkyl chains and β-CD in water (Fig. S2). However, as the OA molar fraction was increased from 13.3% to 33.3%, P(AAm-co-OA) precipitated in water. These results indicated that P(AAm-co-OA) with high OA content formed stable hydrophobic associations instead of host–guest interactions.
a Synthesis and phase transition mechanism of β-CD/OA hydrogels. b Temperature-dependent ATR-FTIR spectra of β-CD/OA hydrogels. c Synchronous and asynchronous 2D correlation spectra of β-CD/OA hydrogels. d Raman spectra of β-CD/OA hydrogels at 20 and 70 °C. e 2D Raman images of the β-CD/OA hydrogels (Intensities of C–H stretching vibration modes).
To help understand the influence of OA molar fraction on the hydrogel networks, the appearance of β-CD/OA organogels before and after water exchange are shown in Fig. S3. As the molar fraction of OA increased from 6.7% to 26.7%, β-CD/OA organogels became transparent due to the high solubility of OA moiety in DMF. The β-CD/OA organogel with an OA molar fraction of 33.3% was translucent, which could be attributed to the high content of β-CD in polymeric networks. After water exchange, only the β-CD/OA hydrogel with an OA molar fraction of 6.7% was transparent, revealing the formation of host–guest interactions in hydrogel networks. As the molar fraction of OA increased from 13.3% to 33.3%, the hydrogels gradually became opaque, indicating that hydrophobic associations were dominant in hydrogel networks. To obtain hydrogels with a high modulus for inflation and then a reduced modulus after phase transition, the hydrogel needed to form at first a strongly hydrophobic crosslinked network after solvent exchange. During the exchange process, water permeated into organogel networks, resulting in the opaque appearance of hydrogels due to hydrophobic association among the OA moieties (Fig. S4).
To support the proposed phase transition behavior of β-CD/OA hydrogels, we performed temperature-dependent FTIR from 20 to 80 °C (Fig. 2b). We focused on the C–H stretching region of methylene groups in OA moieties, including symmetric (νs) and asymmetric stretching vibrations (νas). As the temperature increased, the spectral intensities of methylene groups gradually increased, while the peaks for νas (C–H) and νs (C–H) shifted from 2926 to 2930 cm−1 and from 2853 to 2860 cm−1, respectively. These results suggested that the hydrophobic associations among OA moieties converted into host-gust interaction between OA alkyl groups and β-CD. To extract further information about thermal-induced interaction changes in β-CD/OA hydrogel, we used two-dimensional correlation spectroscopy (2D COS). As shown in Fig. 2c, synchronous and asynchronous spectra were generated to reflect the synchronized and unsynchronized changes of intensities at two given wavenumbers, respectively.33 According to Noda’s judging rule34, the responsive order of the OA moiety to temperature increase is vs(C–H) (methylene groups in hydrophobic association, 2849 cm−1) → vas(C–H) (methylene groups in hydrophobic association, 2916 cm−1) → v(C–H) (methyl groups, 2955 cm−1) → vs(C–H) (methylene groups in β-CD/OA complex, 2857 cm−1) → vas(C–H) (methylene groups in β-CD/OA complex, 2929 cm−1) (Table S1). This sequence revealed that the thermal-induced phase transition of the β-CD/OA hydrogel was driven by the dissociation of hydrophobic interaction.
The thermal-induced phase transition behavior was further evaluated by the Raman spectra of β-CD/OA hydrogels at different temperatures. As shown in Fig. 2d, a new characteristic peak of the methylene groups in OA moieties appeared at 2960 cm−1 after heating from 20 °C to 70 °C, indicating that the chemical environment surrounding OA alkyl chain changed in hydrogel networks. Moreover, the broad peak for the breathing vibration of the glucopyranose ring at 844 cm−1 shifted to a sharp peak at 806 cm−1, indicating that the conformation of β-CD changed after the formation of the host-guest complex, and the vibration of β-CD in the hydrophobic cavity was restricted by the OA. In correlation mode, 2D Raman images of hydrogels showed that hydrophobic interactions (green regions) at 20 °C converted into host-guest interaction (red regions) at 70 °C (Fig. 2e). Moreover, the formation of β-CD/OA complex in hydrogel networks was confirmed by the appearance of an exothermic peak at 70 °C in the DSC curve (Fig. S5), because the host-guest interaction is an enthalpy-dominated binding reaction35,36.
Unlike the actuation speeds of traditional upper critical solution temperature hydrogels are limited by the diffusion rate of water, β-CD/OA hydrogels underwent phase transition (from hydrophobic association to host–guest interaction) without water exchange, with water content being maintained at ~50 wt% during the heating process from 20 to 80 °C (Fig. S6). The rapid phase transition of β-CD/OA hydrogels could be observed in both water and oil at 70 °C (Fig. S7 and Movie S2). When the temperature increased from 20 to 70 °C, the appearance of hydrogel changed from opaque (microphase separation) to transparent (homogeneous state), indicating that the hydrophobic association converted to host–guest interaction in hydrogel networks.
The microphase separation structure of β-CD/OA hydrogels endowed them with high elastic modulus and tensile strength. After heating to 70 °C, the formation of β-CD/OA complexes reduced the cross-linking density of the hydrogel network, resulting in a sharp decrease in the elastic modulus. Therefore, β-CD/OA hydrogel exhibited superior flexibility at 70 °C in comparison to that at 20 °C (Fig. 3a). To increase the modulus gap of β-CD/OA hydrogels before and after heating, we used tunicate cellulose nanocrystals (TCNCs) as reinforcements to enhance the mechanical properties of hydrogels. As the TCNC content reached 0.25 wt%, the elastic modulus of β-CD/OA hydrogels increased from 0.27 MPa to 0.56 MPa due to the high elastic modulus and aspect ratio of TCNCs (Fig. 3b and Fig. S8a)37. On the other hand, the mechanical performance of β-CD/OA hydrogels also depended on the molar fraction of OA that determined the interaction of alkyl chains in hydrogel networks (Fig. 3c and Fig. S8b). When the molar fraction of OA was 6.7%, the elastic modulus of β-CD/OA hydrogels was only 0.015 MPa because the formation of host-guest interactions decreased the crosslinking density of hydrogel networks. As the molar fraction of OA increased to 26.7%, the gap in modulus before and after the phase transition of the hydrogel reached 0.42 MPa, which was attributed to the formation of a densified microphase separation structure at 20 °C. But as the OA molar fraction increased still further to 33.3%, the elastic modulus of hydrogels decreased to 0.34 MPa, due to the conversion of microphase separation to macroscopic phase separation in hydrogel networks.
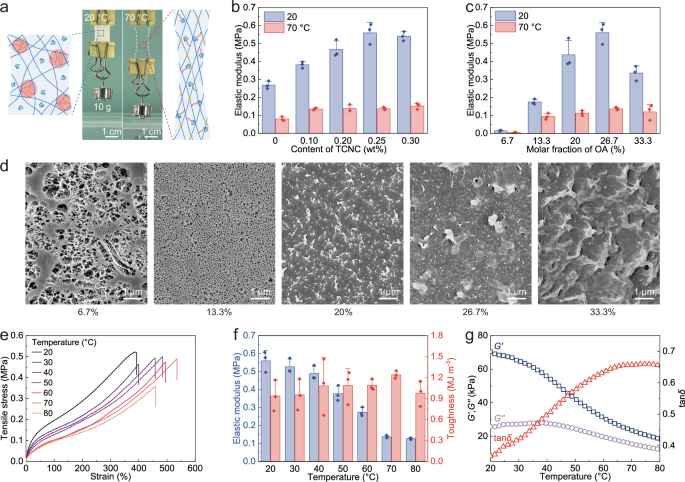
a Photographs and corresponding network structures of β-CD/OA hydrogel under 10 g load at 20 and 70 °C. b The elastic modulus of β-CD/OA hydrogel with different TCNC content and c molar fraction of OA. d SEM images of β-CD/OA hydrogel with different molar fractions of OA. e Stress–strain curves of β-CD/OA hydrogel under different temperatures. f Elastic modulus and toughness of β-CD/OA hydrogel under different temperatures. g The temperature dependence of storage modulus (G′), loss modulus (G″), and loss factor (tan δ) for β-CD/OA hydrogel. Note: for all boxplots, error bars represent standard deviation, n = 3 independent replicates.
As shown in Fig. 3d, a large porous structure (pore size: ~230 nm) could be observed on the surface of the hydrogel with an OA molar fraction of 6.7%, indicating a low crosslinking density. As the molar fraction of OA gradually increased to 13.3%, the pore size of hydrogel significantly decreased to ~63 nm, indicating increased crosslinking density (Fig. S9). As the molar fraction of OA exceeded 20%, the surface of hydrogel became dense and compact, which could be attributed to the formation of hydrophobic associations in hydrogel networks. Finally, at an OA molar fraction of 33.3%, the hydrogel apparently underwent a macroscopic phase separation, leading to a decay of the elastic modulus. Therefore, TCNC content of 0.25 wt% and OA molar fraction of 26.7% were selected for the preparation of β-CD/OA hydrogels, even though the hydrogel showed insignificant transparency at 70 °C (Fig. S3).
The stress-strain curves of β-CD/OA hydrogels under different temperatures are shown in Fig. 3e. The modulus of hydrogels decreased from 0.56 to 0.13 MPa when the temperature increased from 20 to 80 °C, indicating that the dissociation of hydrophobic interactions reduced the crosslinking density of hydrogel networks. The toughness of hydrogel reached a maximum of 1.24 MJ m−3 at 70 °C (Fig. 3f), which is the actuation temperature selected in subsequent experiments. Finally, rheological results showed that both G′ and G″ values of hydrogel decreased as the increase of temperature and tan δ reached a maximum value around 70 °C (Fig. 3g), revealing the possibility of inflating the β-CD/OA hydrogel actuator through this thermal-induced softening process.
An inflatable β-CD/OA hydrogel actuator was obtained by pouring the precursor solution into a tubular mold for polymerization. To optimize the radial expansion of the hydrogel actuator, we first modulated the dimensions of the tubular actuator, including length and thickness. Axial and radial expansion strain–volume relationships of actuators with different lengths were investigated (Fig. S10). All actuators exhibited higher expansion strains in the radical direction in comparison to the axial direction, and the radial expansion strains significantly increased as the temperature rose from 20 to 70 °C, due to the low elastic modulus and high toughness of actuators at 70 °C. Similarly, actuators with different thicknesses exhibited the same expansion trend in both radial and axial directions at 20–70 °C (Fig. S11). According to the above results, we selected an actuator with a thickness of 0.9 mm and a length of 6 mm for further study, as its radial expansion strain was optimal for a given inflation volume.
To obtain inflatable PAMs, the relationship between the expansion behavior of the hydrogel actuators and the pre-pressurized volume (ΔV, the volume of compressed air) was monitored. The strain–ΔV curves of the hydrogel actuators were S-shaped, indicating three stages of the hydrogel actuators during the pre-pressurization process (Fig. 4a). The hydrogel actuator hardly expanded at first, then expanded dramatically and eventually stabilized gradually. Corresponding to the pressure–ΔV curves, the hydrogel actuators generated slight expansion strains until ΔV reached critical pressures, at which point the hydrogel actuator expanded dramatically with the decrease of pressure after exceeding the critical pressure (Fig. 4b). To achieve rapid actuation of the hydrogel actuator switching between two bistable states, the actuator first needed to be pre-pressurized to a high-pressure stable state (stable state I) prior to the critical pressure. To achieve a high actuation strain of the hydrogel actuator, ΔV should be increased as much as possible, which requires a large V0 value. However, when the V0 value exceeded 6 mL, the hydrogel actuator ruptured soon after reaching the critical pressure.
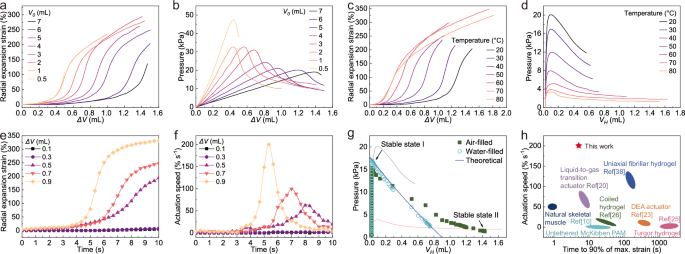
a Radial expansion performance of hydrogel actuators with different initial chamber volumes (V0). b Pressure changes in the inflation process of actuators at different V0. c Radial expansion strain-ΔV curves of hydrogel actuators at different temperatures. d Pressure–volume (VH, hydrogel volume) curves of hydrogel actuators at different temperatures. e Actuation kinetics of actuators inflating at 70 °C with different ΔV. f Actuation speed of hydrogel actuators inflating with different ΔV. g Pressure-volume and theoretical prediction curves of actuators when filled with air and water. h Actuation speed and time to reach 90% of the maximum strain compared to state-of-the-art untethered PAMs10,20,23,25,26,38.
Radial expansion strain–ΔV curves of hydrogel actuators (V0: 6 mL) at different temperatures are shown in Fig. 4c. These results indicated that the radial expansion strains of hydrogel actuators increased with the increase in temperature, reaching a maximum of 347% at 70 °C. Moreover, we calculated the expansion volume (VH) of the hydrogel actuator to establish the pressure-VH relationship at different temperatures (Fig. 4d). Initially, the change of pressure in the curves was linear with pre-pressurization of the hydrogel actuator. After reaching stable state I, the pressure declined, which was attributed to the expansion of the hydrogel actuator by harnessing the snap-through instability. The expansion principle of the hydrogel actuator started with pressurization at 20 °C to reach a high-pressure stable state close to the critical pressure, after which the actuator was separated from the pump and compressor. When the temperature was then increased from 20 to 80 °C, the critical pressure sharply decreased from 20.1 to 3.4 kPa, revealing that the elastic modulus of the β-CD/OA hydrogel could be switched by changing temperature. As the temperature increased to 70 °C, the pressure–VH curve shifted to a lower pressure level due to the decrease in modulus of the hydrogel. The pressure in the untethered PAM then exceeded the decreased critical pressure and triggered the snap-through instability, which caused the hydrogel actuator to expand until the pressure reached stable state II in the pressure–VH curve at 70 °C.
The inflated hydrogel actuators were capable of rapid expansion by harnessing the snap-through instability triggered at 70 °C. The actuation kinetics of the inflated hydrogel actuators with different ΔV are shown in Fig. 4e. The radial expansion strain of hydrogel actuators increased with the increase of ΔV, while all curves of the hydrogel actuator were S-shaped (Fig. S12a), indicating that there was a rapid deformation stage during the actuation process corresponding to the peak of the actuation speed. The actuation speed of the hydrogel actuator significantly increased with increasing ΔV values (Fig. 4f). When ΔV was 0.9 mL, the actuation speed of the hydrogel actuator reached 200% s−1. Since the pressure and volume of a given number of gas molecules in the system were inversely proportional, the pressure of the hydrogel actuator concomitantly decreased from 14.6 to 2.3 kPa within 10 s and finally stabilized at 1.3 kPa. (Fig. S12b). The relationship between the volume and pressure of the hydrogel actuator (ΔV, 0.9 mL) during the expansion process is shown in Fig. 4g. The final VH of the hydrogel actuator was 1.43 mL, which was 50 times the initial VH (0.026 mL).
To clarify the actuation mechanism of our PAM, we compared the measured pressure–VH curve to the theoretical curve calculated by the ideal gas equation. We found that the experimental data deviated from the ideal gas equation, resulting in a larger final VH. To explain this phenomenon, we hypothesized that the hydrogel actuator could be powered by both thermal expansion of the gas inside the actuator and evaporation of water from its internal surface of hydrogel actuator. To test this hypothesis, we conducted a control experiment by replacing the air in the hydrogel actuator with an equal amount of water and retaining the air in the chamber during actuation. In this case, the pressure–VH curve of the water-filled hydrogel actuator was consistent with the theoretical prediction, indicating that the expansion of the hydrogel was driven only by the snap-through instability. But in general, we should consider the thermal expansion of the air in the hydrogel actuator at 70 °C (1.17 times that at 20 °C). Therefore, the actuation of our PAM was driven by a combination of properties afforded by the hydrogel actuator; namely snap-through instability, thermal expansion of trapped air, and water evaporation from the inner surface.
The actuation speed and the time to reach 90% maximum strain of our PAMs were compared to those of natural muscles and reported works (Fig. 4h). Liquid-to-gas transition-based PAMs20 and untethered McKibben PAMs10 could complete the expansion and contraction, respectively, after more than ten seconds, but the actuation speed was limited by the lack of a bistable mechanism. The dielectric elastomer actuator exhibited bistable deformation under electrical stimulation, but the inherent viscosity of the dielectric elastomers limited their expansion speed under applied voltage23. In addition, the system required the connection of a chamber 5–10 times the volume of the actuator, restricting its integrated application. The solvent-responsive coil-shaped hydrogels showed large actuation strain, but their actuation speed was still limited by the permeation speed of solvent molecules26. The selectively permeable membrane enabled the turgor hydrogel a huge expansion actuation force, but was not conducive to the penetration rate of water molecules25. Uniaxial fibrillar hydrogel actuators improved actuation speed by reducing the permeation path of water (130% s−1), which decreased sharply with the increase of swelling ratio, resulting in a great extension of the actuation time.38 Although the actuation speed of natural skeletal muscles was lower than that of our hydrogel actuators, it took less time to complete the actuation because the actuation strain of the muscles was typically only about 20%. Benefiting from the combined effect of the snap-through instability, thermal expansion of the gas inside the actuator, and evaporation of water on the internal surface of the hydrogel actuator, our untethered PAM achieved an actuation strain speed of up to 200% s−1. In addition, the compressibility and flowability of air enabled the hydrogel actuator to quickly equalize air pressure within the untethered PAM.
For the miniaturization of the untethered PAM, we reduced the volume of the air chamber (V0) connected to the hydrogel. When ΔV was fixed at 0.3 mL, decreasing V0 could significantly improve the actuation strain and speed of the hydrogel actuator (Fig. 5a). With V0 of 1 mL, the radial expansion strain and the actuation speed of the hydrogel actuator reached 210% and 163% s−1, respectively (Fig. S13). The actuation force of the hydrogel actuator at different expansion strains is shown in Fig. 5b. The blocking actuation force (Actuation strain: 0%) reached up to 0.45 N after heating for 3 s, and then the actuation force decreased with increasing expansion strain. Therefore, the untethered PAM with a V0 of 1 mL enabled the miniaturization and integrated applications of the hydrogel actuator with a work capacity of 27.2 J kg−1.
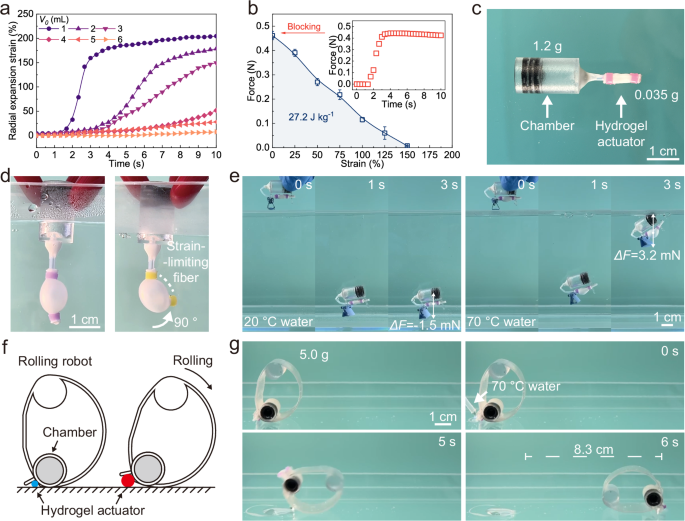
a Actuation kinetics of miniaturized hydrogel actuator inflation at 70 °C with different V0. b Actuation force of hydrogel actuator with different actuation strains. Error bars represent standard deviation, n = 3 independent replicates. c Untethered PAM consisting of a chamber and a hydrogel actuator. d The expansion and bending of the hydrogel actuator. e The diving robot sinking in 20 °C water and floating up in 70 °C water, respectively. f Schematic design of an integrated rolling robot. g Photographs of the rolling process of the robot triggered by 70 °C water.
For applications, our miniaturized PAM consisted of a 3D-printed chamber (polyethylene terephthalate glycol, PETG) and a hydrogel actuator (Fig. 5c). The miniaturized PAM could expand and bend (assisted by strain-limiting optical fibers) rapidly in 70 °C water (Fig. 5d and Movie S3). One application we explored was to use the untethered PAM as a diving robot to detect water temperature by adjusting its buoyancy underwater (Fig. 5e and Movie S4). The diving robot quickly sank to the bottom once it was placed into 20 °C water due to the gravity it experienced being greater than its own buoyancy, and the resultant force was −1.5 mN. However, when placed in 70 °C water, the diving robot first sank to the bottom but then floated back up to the surface within 2 s, due to the thermal-induced expansion of the hydrogel actuator, and the resultant force increased to 3.2 mN. The miniaturized design made the gravity of the entire robot less than the buoyancy created by the expansion of the hydrogel, a buoyancy that overcame the limitation of bulky equipment such as compressors. The ability to regulate buoyancy enabled hydrogel actuators to be used in potential areas such as bionic fish, diving equipment, and underwater rescue. Another application we explored, inspired by the germination behavior of seeds that expand and push away soil and stones, we designed a rolling robot by using the untethered PAM. The rolling robot comprised an integrated chamber, wheels (PETG), and a hydrogel actuator with a total weight of 5 g (Fig. 5f). The expansion of the hydrogel actuator changed the position of the gravity center of the robot, resulting in a rolling motion. The hydrogel actuator could propel the rolling robot (140 times its own weight) forward 8.3 cm within 6 s under thermal stimulation (Fig. 5g and Movie S5). Our strategy gives intelligent robots a highly integrated architecture and environmental adaptability, enabling portable, miniaturized mobility and transportation.